DeepMind’s AlphaFold 2 reveal: Convolutions are out, attention is in
 ]
]
DeepMind, the AI unit of Google that invented the chess champ neural network AlphaZero a few years back, shocked the world again in November with a program that had solved a decades-old problem of how proteins fold. The program handily beat all competitors, in what one researcher called a “watershed moment” that promises to revolutionize biology.
AlphaFold 2, as it’s called, was described at the time only in brief terms, in a blog post by DeepMind and in a paper abstract provided by DeepMind for the competition in which they submitted the program, the Critical Assessment of Techniques for Protein Structure Prediction biannual competition.
Last week, DeepMind finally revealed just how it’s done, offering up not only a blog post but also a 16-page summary paper written by DeepMind’s John Jumper and colleagues in Nature magazine, a 62-page collection of supplementary material, and a code library on GitHub. A story on the new details by Nature’s Ewan Calloway characterizes the data dump as “protein structure coming to the masses.”
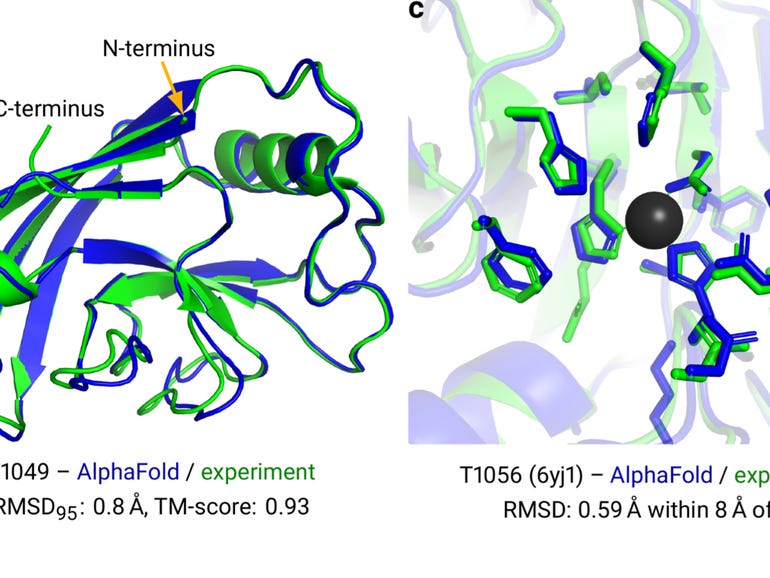
So, what have we learned? A few things. As the name suggests, this neural net is the successor to the first AlphaFold, which had also trounced competitors in the prior competition in 2018. The most immediate revelation of AlphaFold 2 is that making progress in artificial intelligence can require what’s called an architecture change.
The architecture of a software program is the particular set of operations used and the way they are combined. The first AlphaFold was made up of a convolutional neural network, or “CNN,” a classic neural network that has been the workhorse of many AI breakthroughs in the past decade, such as containing triumphs in the ImageNet computer vision contest.
But convolutions are out, and graphs are in. Or, more specifically, the combination of graph networks with what’s called attention.
A graph network is when some collection of things can be assessed in terms of their relatedness and how they’re related via friendships – such as people in a social network. In this case, AlphaFold uses information about proteins to construct a graph of how near to one another different amino acids are.
Also: Google DeepMind’s effort on COVID-19 coronavirus rests on the shoulders of giants
These graphs are manipulated by the attention mechanism that has been gaining in popularity in many quarters of AI. Broadly speaking, attention is the practice of adding extra computing power to some pieces of input data. Programs that exploit attention have lead to breakthroughs in a variety of areas, but especially natural language processing, as in the case of Google’s Transformer.
The part that used convolutions in the first AlphaFold has been dropped in Alpha Fold 2, replaced by a whole slew of attention mechanisms.
Use of attention runs throughout AlphaFold 2. The first part of AlphaFold is what’s called EvoFormer, and it uses attention to focus processing on computing the graph of how each amino acid relates to another amino acid. Because of the geometric forms created in the graph, Jumper and colleagues refer to this operation of estimating the graph as “triangle self-attention.”
DeepMind / Nature
Echoing natural language programs, the EvoFormer allows the triangle attention to send information backward to the groups of amino acid sequences, known as “multi-sequence alignments,” or “MSAs,” a common term in bioinformatics in which related amino acid sequences are compared piece by piece.
The authors consider the MSAs and the graphs to be in a kind of conversation thanks to attention – what they refer to as a “joint embedding.” Hence, attention is leading to communication between parts of the program.
DeepMind / Nature
The second part of AlphaFold 2, following the EvoFormer, is what’s called a Structure Module, which is supposed to take the graphs that the EvoFormer has built and turn them into specifications of the 3-D structure of the protein, the output that wins the CASP competition.
Here, the authors have introduced an attention mechanism that calculates parts of a protein in isolation, called an “invariant point attention” mechanism. They describe it as “a geometry-aware attention operation.”
The Structure Module initiates particles at a kind of origin point in space, which you can think of as a 3-D reference field, called a “residue gas,” and then proceeds to rotate and shift the particles to produce the final 3-D configuration. Again, the important thing is that the particles are transformed independently of one another, using the attention mechanism.
DeepMind / Nature
Why is it important that graphs, and attention, have replaced convolutions? In the original abstract offered for the research last year, Jumper and colleagues pointed out a need to move beyond a fixation on what are called “local” structures.
Going back to AlphaFold 1, the convolutional neural network functioned by measuring the distance between amino acids, and then summarizing those measurements for all pairs of amino acids as a 2-D picture, known as a distance histogram, or “distogram.” The CNN then operated by poring over that picture, the way CNNs do, to find local motifs that build into broader and broader motifs spanning the range of distances.
But that orderly progression from local motifs can ignore long-range dependencies, which are one of the important elements that attention supposedly captures. For example, the attention mechanism in the EvoFormer can connect what is learned in the triangle attention mechanism to what is learned in the search of the MSA – not just one section of the MSA, but the entire universe of related amino acid sequences.
Hence, attention allows for making leaps that are more “global” in nature.
Another thing we see in AlphaFold is the end-to-end goal. In the original AlphaFold, the final assembly of the physical structure was simply driven by the convolutions, and what they came up with.
In AlphaFold 2, Jumper and colleagues have emphasized training the neural network from “end to end.” As they say:
“Both within the Structure Module and throughout the whole network, we reinforce the notion of iterative refinement by repeatedly applying the final loss to outputs then feeding the outputs recursively to the same modules. The iterative refinement using the whole network (that we term ‘recycling’ and is related to approaches in computer vision) contributes significantly to accuracy with minor extra training time.”
Hence, another big takeaway from AlphaFold 2 is the notion that a neural network really needs to be constantly revamping its predictions. That is true both for the recycling operation, but also in other respects. For example, the EvoFormer, the thing that makes the graphs of amino acids, revises those graphs at each of the multiple stages, what are called “blocks,” of the EvoFormer. Jumper and team refer to this constant updates as “constant communication” throughout the network.
As the authors note, through constant revision, the Structure piece of the program seems to “smoothly” refine its models of the proteins. “AlphaFold makes constant incremental improvements to the structure until it can no longer improve,” they write. Sometimes, that process is “greedy,” meaning, the Structure Module hits on a good solution early in its layers of processing; sometimes, it takes longer.
Also: AI in sixty seconds
In any event, in this case the benefits of training a neural network – or a combination of networks – seem certain to be a point of emphasis for many researchers.
Alongside that big lesson, there is an important mystery that remains at the center of AlphaFold 2: Why?
Why is it that proteins fold in the ways they do? AlphaFold 2 has unlocked the prospect of every protein in the universe having its structure revealed, which is, again, an achievement decades in the making. But AlphaFold 2 doesn’t explain why proteins assume the shape that they do.
Proteins are amino acids, and the forces that make them curl up into a given shape are fairly straightforward – things like certain amino acids being attracted or repelled by positive or negative charges, and some amino acids being “hydrophobic,” meaning, they stay farther away from water molecules.
What is still lacking is an explanation of why it should be that certain amino acids take on shapes that are so hard to predict.
AlphaFold 2 is a stunning achievement in terms of building a machine to transform sequence data into protein models, but we may have to wait for further study of the program itself to know what it is telling us about the big picture of protein behavior.
兩大頂級 AI 演算法開源!Alphafold2 蛋白質預測準度逼近滿分,將顛覆生醫產業樣貌
 ]
]
【為什麼我們要挑選這篇文章】蛋白質結構預測是生物學的大難題。在過去的蛋白質結構預測比賽中,最優秀的團隊僅能從滿分 100 中取得 40 分。但到了 2020 年,AI 系統 Alphafold 2 取得高達 92.4 分的好成績,解決了蛋白質的折疊問題。近日 Alphafold 2 開源,將會掀起生醫界的波瀾。(責任編輯:郭家宏)
本文經 AI 新媒體量子位(公眾號 ID:QbitAI)授權轉載,轉載請連繫出處
作者:量子位
喜大普奔!近日一波 Nature、Science 齊發文,可把學術圈的人們高興壞了。
(TO 編按:喜大普奔為中國網路用語,是喜文樂見、大快人心、普天同慶、奔走相告的縮寫)
一邊是「AI 界年度十大突破」AlphaFold2 終於終於開源,登上 Nature。
另一邊 Science 又出報導:華盛頓大學竟然還搞出了一個比 AlphaFold2 更快更輕便的演算法,只需要一個 NVIDIA RTX2080 GPU,10 分鐘就能算出蛋白質結構!
要知道,當年 AlphaFold2 橫空出世,那是真沸騰了學術圈。
不僅 Google CEO 皮蔡、馬斯克、李飛飛等大 V(微博上有眾多粉絲的人物)紛紛按讚,連馬普所的演化生物研究所所長 Andrei Lupas 都直言:它會改變一切。
結構生物學家 Petr Leiman 感嘆,我用價值一千萬美元的電子顯微鏡努力地解了好幾年,Alphafold2 竟然一下就算出來了。
更是有生物學網友表示絶望,感覺專業前途渺茫。
而今天這一波 Nature、Science 神仙打架,再次點燃話題度。
Alphafold2 解決蛋白質摺疊預測問題,能加速新藥開發
先說被頂刊爭相報導的 Alphafold2,它作為一個 AI 模型,為何引起各界狂熱?
因為它一出來,就解決了生物學界最棘手的問題之一。這個問題於 1972 年被克里斯蒂安.安芬森提出,它的驗證曾經困擾科學家 50 年:
「給定一個胺基酸序列,理論上就能預測出蛋白質的 3D 結構。」
蛋白質由胺基酸序列組成,但真正決定蛋白質作用的,是它的 3D 結構,也就是胺基酸序列的摺疊方式。
為了驗證這個理論,科學家們嘗試了各種手段,但在 CASP14(蛋白質結構預測比賽)中,準確性也只達到 40 分左右(滿分 100)。
直到去年 12 月,Alphafold2 出現,將這一準確性直接拔高到了 92.4/100,和蛋白質真實結構之間只差一個原子的寬度,真正解決了蛋白質摺疊的問題。
Alphafold2 於當年入選 Science 年度十大突破,被稱作結構生物學「革命性」的突破、蛋白質研究領域的里程碑。
它的出現,能更好地預判蛋白質與分子結合的機率,從而極大地加速新藥研發的效率。
今天,Alphafold2 的開源,又進一步在 AI 和生物學界激起了一大波浪。
Google CEO 皮蔡很高興。
亦有生物學博士表示:未來已來!
來自 UC 柏克萊 AI 實驗室的博士 Roshan Rao 看過後表示,這份程式碼碼看起來不僅容易使用,而且文檔也非常完善。
現在,是時候藉著這份開源算法,弄清 Alphafold2 的魔術是怎麼變的了。
AlphaFold2 使用多序列比對,將蛋白質結構整合到演算法
研究人員強調,這是一個完全不同於 AlphaFold 的新模型。
2018 年的 AlphaFold 使用的神經網絡是類似 ResNet 的殘差卷積網絡,到了 AlphaFold2 則借鑒了 AI 研究中最近新興起的 Transformer 架構。
Transformer 使用注意力機制興起於 NLP 領域,用於處理一連串的文字內容序列。
而胺基酸序列正是和文字內容類似的數據結構,AlphaFold2 利用多序列比對,把蛋白質的結構和生物資訊整合到了深度學習演算法中。
AlphaFold2 用初始胺基酸序列與同源序列進行比對,直接預測蛋白質所有重原子的三維坐標。
從模型圖中可以看到,輸入初始胺基酸序列後,蛋白質的基因資訊和結構資訊會在數據庫中進行比對。
多序列比對的目標是使參與比對的序列中有儘可能多的序列具有相同的鹼基,這樣可以推斷出它們在結構和功能上的相似關係。
比對後的兩組訊息會組成一個 48block 的 Evoformer 塊,然後得到較為相似的比對序列。
比對序列進一步組合 8 blocks 的結構模型,從而直接構建出蛋白質的 3D 結構。
最後兩步過程還會進行 3 次循環,可以使預測更加準確。
更快的演算法:RoseTTAFold 用一般電腦就可操作
AlphaFold2 首次公佈的時候並沒有透露太多技術細節。
在華盛頓大學,同樣致力於蛋白質領域的 David Baker 一度陷入失落:
如果有人已經解決了你正在研究的問題,但沒有透露他們是如何解決的,你該如何繼續研究?
不過他馬上重整旗鼓,帶領團隊嘗試能不能復現 AlphaFold2 的成功。
幾個月後,Baker 團隊的成果不僅在準確度上和 AlphaFold2 不相上下,還在計算速度和算力需求上實現了超越。
就在 AlphaFold2 開源論文登上 Nature 的同一天,Baker 團隊的 RoseTTAFold 也登上 Science。
RoseTTAFold 只需要一塊 RTX2080 顯卡,就能在 10 分鐘左右計算出 400 個胺基酸殘基以內的蛋白質結構。
這樣的速度,意味著什麼?
那就是研究蛋白質的科學家不用再排隊申請超級電腦運算資源了,小型團隊和個人研究者只需要一台普通的個人電腦就能輕鬆展開研究。
RoseTTAFold 的秘訣在於採用了 3 軌注意力機制,分別關注蛋白質的一級結構、二級結構和三級結構。
再通過在三者之間加上多處連接,使整個神經網絡能夠同時學習 3 個維度層次的訊息。
考慮到現在市場上顯卡不太好買,Baker 團隊還貼心的搭建了公共伺服器,任何人都可以提交蛋白質序列並預測結構。
自伺服器建立以來,已經處理了來自全世界研究者提交的幾千個蛋白質序列。
這還沒完,團隊發現如果同時輸入多個胺基酸序列,RoseTTAFold 還可以預測出蛋白質複合體的結構模型。
對於多個蛋白質組成的複合體,RoseTTAFold 的實驗結果是在 24 GB 顯存的 NVIDIA Titan RTX 上計算 30 分鐘左右。
現在整個網絡是用單個胺基酸序列訓練的,團隊下一步計劃用多序列重新訓練,在蛋白質複合體結構預測上還可能有提升空間。
正如 Baker 所說:
我們的成果可以幫助整個科學界,為生物學研究加速。
Alphafold2 開源 地址
RoseTTAFold 開源 地址
相關論文:Nature、Science
參考連結:TechCrunch、Nature
延伸閱讀
• DeepMind 巨虧 180 億、加拿大獨角獸遭 3 折賤賣,AI 公司為何難有「好下場」?
• 【生物界的 AlphaGo 更強大】DeepMind 宣布 AI 能成功預測蛋白質結構,有機會翻轉未來醫療面貌
• 【不治之症將有解?】AI 篩選潛力藥物分子,四週內生成「超完美新藥」!
(本文經 AI 新媒體量子位 授權轉載,並同意 TechOrange 編寫導讀與修訂標題,原文標題為 〈两大顶级 AI 算法一起开源!Nature、Science 齐发 Alphafold2 相关重磅,双厨狂喜~〉。首圖來源:GitHub)
訂閱《TechOrange》每日電子報!
DeepMind open-sources AlphaFold 2 for protein structure predictions
 ]
]
All the sessions from Transform 2021 are available on-demand now. Watch now.
Let the OSS Enterprise newsletter guide your open source journey! Sign up here.
DeepMind this week open-sourced AlphaFold 2, its AI system that predicts the shape of proteins, to accompany the publication of a paper in the journal Nature. With the codebase now available, DeepMind says it hopes to broaden access for researchers and organizations in the health care and life science fields.
The recipe for proteins — large molecules consisting of amino acids that are the fundamental building blocks of tissues, muscles, hair, enzymes, antibodies, and other essential parts of living organisms — are encoded in DNA. It’s these genetic definitions that circumscribe their three-dimensional structures, which in turn determine their capabilities. But protein “folding,” as it’s called, is notoriously difficult to figure out from a corresponding genetic sequence alone. DNA contains only information about chains of amino acid residues and not those chains’ final form.
In December 2018, DeepMind attempted to tackle the challenge of protein folding with AlphaFold, the product of two years of work. The Alphabet subsidiary said at the time that AlphaFold could predict structures more precisely than prior solutions. Its successor, AlphaFold 2, announced in December 2020, improved on this to outgun competing protein-folding-predicting methods for a second time. In the results from the 14th Critical Assessment of Structure Prediction (CASP) assessment, AlphaFold 2 had average errors comparable to the width of an atom (or 0.1 of a nanometer), competitive with the results from experimental methods.
AlphaFold draws inspiration from the fields of biology, physics, and machine learning. It takes advantage of the fact that a folded protein can be thought of as a “spatial graph,” where amino acid residues (amino acids contained within a peptide or protein) are nodes and edges connect the residues in close proximity. AlphaFold leverages an AI algorithm that attempts to interpret the structure of this graph while reasoning over the implicit graph it’s building using evolutionarily related sequences, multiple sequence alignment, and a representation of amino acid residue pairs.
In the open source release, DeepMind says it significantly streamlined AlphaFold 2. Whereas the system took days of computing time to generate structures for some entries to CASP, the open source version is about 16 times faster. It can generate structures in minutes to hours, depending on the size of the protein.
Real-world applications
DeepMind makes the case that AlphaFold, if further refined, could be applied to previously intractable problems in the field of protein folding, including those related to epidemiological efforts. Last year, the company predicted several protein structures of SARS-CoV-2, including ORF3a, whose makeup was formerly a mystery. At CASP14, DeepMind predicted the structure of another coronavirus protein, ORF8, that has since been confirmed by experimentalists.
Beyond aiding the pandemic response, DeepMind expects AlphaFold will be used to explore the hundreds of millions of proteins for which science currently lacks models. Since DNA specifies the amino acid sequences that comprise protein structures, advances in genomics have made it possible to read protein sequences from the natural world, with 180 million protein sequences and counting in the publicly available Universal Protein database. In contrast, given the experimental work needed to translate from sequence to structure, only around 170,000 protein structures are in the Protein Data Bank.
DeepMind says it’s committed to making AlphaFold available “at scale” and collaborating with partners to explore new frontiers, like how multiple proteins form complexes and interact with DNA, RNA, and small molecules. Earlier this year, the company announced a new partnership with the Geneva-based Drugs for Neglected Diseases initiative, a nonprofit pharmaceutical organization that used AlphaFold to identify fexinidazole as a replacement for the toxic compound melarsoprol in the treatment of sleeping sickness.
Highly accurate protein structure prediction with AlphaFold
![]() ]
]
Proteins are essential to life, and understanding their structure can facilitate a mechanistic understanding of their function. Through an enormous experimental effort1–4, the structures of around 100,000 unique proteins have been determined5, but this represents a small fraction of the billions of known protein sequences6,7. Structural coverage is bottlenecked by the months to years of painstaking effort required to determine a single protein structure. Accurate computational approaches are needed to address this gap and to enable large-scale structural bioinformatics. Predicting the 3-D structure that a protein will adopt based solely on its amino acid sequence, the structure prediction component of the ‘protein folding problem’8, has been an important open research problem for more than 50 years9. Despite recent progress10–14, existing methods fall far short of atomic accuracy, especially when no homologous structure is available. Here we provide the first computational method that can regularly predict protein structures with atomic accuracy even where no similar structure is known. We validated an entirely redesigned version of our neural network-based model, AlphaFold, in the challenging 14th Critical Assessment of protein Structure Prediction (CASP14)15, demonstrating accuracy competitive with experiment in a majority of cases and greatly outperforming other methods. Underpinning the latest version of AlphaFold is a novel machine learning approach that incorporates physical and biological knowledge about protein structure, leveraging multi-sequence alignments, into the design of the deep learning algorithm.
How DeepMind’s AI Cracked a 50-Year Science Problem Revealed
 ]
]
Source: Saylowe/Pixabay
DeepMind, a Google-owned artificial intelligence (AI) company based in the United Kingdom, made scientific history when it announced last November that it had a solution to a 50-year-old grand challenge in biology—protein folding. This AI machine learning breakthrough may help accelerate the discovery of new medications and novel treatments for diseases. On July 15, 2021 DeepMind revealed details on how its AI works in a new peer-reviewed paper published in Nature, and made its revolutionary AlphaFold version 2.0 model available as open-source on GitHub.
The three-dimensional (3D) shape and function of proteins are determined by the sequence of its amino acids. AlphaFold predicts three-dimensional (3D) models of protein structures. Open-source software makes code open and available to download, which over time may also help to improve the software as well.
“Predicting the 3-D structure that a protein will adopt based solely on its amino acid sequence, the structure prediction component of the ‘protein folding problem’, has been an important open research problem for more than 50 years,” wrote Demis Hassabis, John Jumper, Richard Evans, and their DeepMind research colleagues who authored the new paper.
According to the DeepMind researchers, the structures of roughly 100,000 unique proteins have already been determined by scientists, however this “represents a small fraction of the billions of known protein sequences.” There are approximately 200 million known proteins in existence, and roughly 30 million new ones are discovered annually.
“Structural coverage is bottlenecked by the months to years of painstaking effort required to determine a single protein structure,” wrote the researchers. “Accurate computational approaches are needed to address this gap and to enable large-scale structural bioinformatics.”
In molecular biology, proteins are large, chemically complex large biomolecules molecules called macromolecules that consist of hundreds or even thousands of amino acids connected in long chains linked through covalent peptide bonds. Amino acids, the building blocks of proteins, are organic molecules that combine to form proteins. There are around 20 types of amino acids in proteins, and each has different chemical traits.
“Here we provide the first computational method that can regularly predict protein structures with atomic accuracy even where no similar structure is known,” reported the DeepMind researchers. “We validated an entirely redesigned version of our neural network-based model, AlphaFold, in the challenging 14th Critical Assessment of protein Structure Prediction (CASP14), demonstrating accuracy competitive with experiment in a majority of cases and greatly outperforming other methods.”
AlphaFold uses input features extracted from templates, amino-acid sequence, and multiple sequence alignments (MSAs) for inference and produces features outputs that include per-residue confidence scores, atom coordinates, and the distogram, which is a histogram of distances between pairs of pixels in an image. A histogram is a graphical display of numerical data that represents a frequency distribution with rectangles or bars where the widths represent class intervals and the areas are proportional to the corresponding frequencies. TensorFlow was used to develop the artificial neural networks and Python was used for the data analysis. Markov Chain Monte Carlo based Bayesian analysis was also used.
Proteins are essential to life, and understanding their structure can facilitate a mechanistic understanding of their function,” the researchers wrote.
DeepMind’s groundbreaking work and the open-sourcing of AlphaFold version 2.0 may help accelerate innovation through scientific collaboration in life sciences, biotechnology, data science, molecular biology, pharmaceutical, and more industries to discover novel treatments and medications to extend human longevity in the future.
Copyright © 2021 Cami Rosso All rights reserved.
線上諮詢
與我們合作,馬上展開全新的創作里程碑
- 📅 立即預約,30 秒完成!
- 🎯 與創辦人 1 對 1 交流,獲得專屬建議! 🎯 與創辦人 1 對 1 交流!